ICCS Portland Plenary Session II:
“Optimization of Multicolor Immunophenotyping Assays: Critical Components and Practical Guidelines”
Chairs: Andrea Illingworth and Rob Sutherland.
1. “Optimizing a PNH Assay” jointly delivered by Andrea Illingworth, Dahl-Chase Diagnostic Services, USA and Rob Sutherland, University Health Network/TGH, Canada.
Dahl-Chase Diagnostic Services has tested 7,126 patient samples to date and of these 6.1% have been positive for GPI deficient cells. Breaking this down into clone sizes, 3.7% were greater than 1%, 0.7% were between 0.1-1%, and 1.7% were less than 0.1%. Clones sizes were generally stable in patients initially seen with a 0.1% or less clone size, whereas there were more changes at a 1% or greater cutoff.
For their RBC assay Ms. Illingworth recommends using CD235a FITC (clone KC16) and CD59 PE (clone MEM43) along with scatter. There is no need to run CD55, as CD59 is sufficient and superior. To keep aggregation of RBC’s below 2%, both CD235a and CD59 may have to be diluted below their saturating concentration. RBC tube should be washed twice after staining and “racked” hard (run vigorously along a tube rack) prior to acquisition to avoid aggregation. Before running, instrument voltages are optimized so unstained RBC’s are on scale and appropriate compensation for these settings are established by running CD235a and CD59 single stained control tubes. A dual stained healthy donor tube is used to check compensation in a two parameter histogram of CD235a versus CD59 and Type I, II and III regions are established using a single parameter histogram gated on the CD235a positive cells. Routinely, they collect 50,000 events and then evaluate the data. If a single type III event is detected, then the tube is rerun and 500,000 to 1,000,000 events are collected. In their RBC assay, background levels are approximately 2 to 6 Type III events per million cells collected.
Rob Sutherland discussed the WBC assay and the reagents and antibodies most commonly used for it. CD55 and CD59 are out; they both lack the sensitivity and specificity required for a reliable PNH assay. FLAER is the most versatile reagent for the detection of PNH clones in WBCs because it binds specifically to the GPI anchor. Consequently, it is both sensitive and specific and can be used to detect GPI deficient clones in both granulocytes and monocytes. In addition to FLAER, CD16, CD66b and CD24 are the more commonly used markers to detect GPI deficient clones in granulocytes, while CD14 is commonly used for monocytes. Recently, Dr. Sutherland and other labs have been looking at CD157 to replace CD14 and possibly CD24 in the granulocyte assay, which he indicated was “looking good”. For high sensitivity assays using forward and side scatter alone or scatter plus CD45 is not acceptable for gating because spurious events that may not represent granulocytes or monocytes might be included in the gate, leading to a false conclusion that a PNH population is present. Instead CD15 in combination with side scatter is recommended as a gating parameter for granulocytes and CD64 versus scatter for monocytes. CD33 is no longer recommended as a gating parameter because of its decreased expression on immature cells and cells from patients with MDS.
Dr. Sutherland also uses stained cells to set compensation, setting each parameter with a single color tube stained with CD3. He noted that the CD3 negative unstained population in each tube serves as its own internal negative control. He also reminded the group that CD3 FITC should not be used to set up compensation for the FLAER reagent, which is an Alexa 488 conjugate.
For analysis of granulocytes, he used a series of gates progressing from forward versus side scatter to eliminate aggregates and debris, then CD45 versus side scatter to exclude unstained cells, followed by a CD15 PECy5 versus side scatter plot to uniquely identify lymphocytes, monocytes and granulocytes. From each of these populations a FLAER versus CD24 PE plot is created. The lymphocyte gated and monocyte gated FLAER versus CD24 dot plots serve as controls while the number of dual FLAER and CD24 negative (GPI deficient) events in the plot gated on granulocytes is reported. For the analysis of PNH clones in the monocyte population a similar strategy is used, replacing CD15 PE-Cy5 with CD64 PE-Cy5 and reporting out the number of dual negative FLAER versus CD14 PE events falling in the histogram gated on the monocyte population.
Both speakers suggested validating the RBC and WBC assays by spiking blood samples with blood from a known PNH sample. By running serial dilutions, both the linearity and sensitivity of the assay can be determined. Background levels are determined running samples from healthy donors and were 0.0013% and 0.0033% for granulocytes and monocytes respectively in the examples shown. The speakers also commented on the need for better proficiency materials and noted that in the absence of a good program, sharing samples among colleagues was better than doing nothing at all. For additional information see Borowitz et al., 2010.
2. “Developing a 10-Color Protocol for the Diagnosis of Leukemia and Lymphoma”, Mike Keeney, London Health Services Centre, Canada.
Dr. Keeney talked about the development of four 10-color tubes that can be used to correctly identify the majority of leukemia and lymphoma cases. One tube each was created to identify T and B cell malignancies and two tubes for myeloid/monocytic cells and their precursors. The monoclonal antibodies (mAbs) used in each and the choice of fluorochromes are listed in Table 1. In addition to these 10 color tubes, he is running as needed two 5 color tubes, one for Hairy cells (CD103 FITC/ CD11c PE/ CD45 PE-TR/CD25 PE-Cy5.5/CD19 PE-Cy7) and one for intracellular antigens (CD34 FITC/MPO PE/CD45 PE-TR/CD79a PE-Cy5.5/CD3 PECy7).
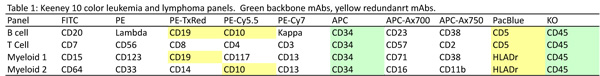
Strategies used in the panel design included:
A. The inclusion of backbone and redundant mAbs within the panels makes it easier to track populations between tubes, merge the data and troubleshoot unexpected results. In this setup, backbone CD45 and CD34 mAbs were included in all tubes, CD5 was included in both lymphocyte tubes, HLADr in both myeloid tubes, and CD10 and CD19 were included in the a lymphoid and myeloid tube.
B. All the mAbs were individually titrated using a large number of cells with a brightly positive population for the marker. Each 10 color cocktail was designed so that a final 100µL volume of cocktail would be added to each tube. If the final cocktail volume required was less than 100µL, PBS was added to make up the difference. Moreover, for consistency all individual mAb tiers were performed by adding 100µL of the diluted mAb. The titer, which gave the maximum signal to noise, was deemed optimal.
C. To reduce crossover and keep all mAbs on scale, those mAbs with extremely high intensity were diluted with the identical clone of unlabeled mAb.
D. Based on preliminary data, cocktails of all mAbs were combined and used within 2 weeks or discarded because of tandem breakdown. Dr. Keeney noted that this practice is controversial and suggested that other investigators and the manufacturers themselves develop guidelines for the storage of cocktails containing tandems.
Instrument setup, data acquisition and analysis.
Initially PMT voltages for each fluorochrome were optimized using CD8. Next appropriate compensation values were determined using CD45. The optimal compensation matrix was determined over 5 days and the average was calculated and used for the final matrix. Daily calibration beads were used, recording the MFI and CV’s for each channel. Using this system, the only time he changes the compensation matrix is following an instrument adjustment such as a PM, repair or realignment.
For staining a standard whole blood method was used, including a wash before addition of mAbs to the B cells tube to avoid distraction of anti-kappa and lambda mAbs by plasma immunoglobulin. The cells were lysed with ammonium chloride containing a 0.25% formaldehyde fixative and washed with PBS containing 1% BSA. Routinely, 100,000 events were collected using a sequential gating strategy starting with forward scatter versus side scatter to exclude dead cells, aggregates and debris, then a singlet gate using forward scatter peak versus area, a CD45 versus side scatter to exclude unstained cells and color code individual leukocyte populations, and finally a run status check using time versus APXAx750.
Analysis of data with 10 different colors is complex with 1024 possible bivariate plots. Dr. Keeney does all analysis post acquisition and starts by visualizing each parameter versus side scatter. The availability of new fluorochromes, coupled with multilaser instruments with simplified setup and compensation makes routine 10-color analysis possible for the clinical laboratory.
3. “Optimizing a Polychromatic Flow Cytometric Assay for the Diagnosis of Hematolymphoid Neoplasia”, Steve Kussick, PhenoPath Laboratories, USA.
Dr. Kussick presented his experience setting up high content, clinical, multiparameter flow panels. To begin development, he chose to create a 9-color T cell panel, rationalizing that the expression patterns of these antigens were well defined and that the information and experience gained from this “simple” tube could be applied to more complex situations. As a very first step the question(s) to be answered by the tube were defined and then the mAbs required to address the question were chosen. The selection of mAbs was guided in part by the 2006 Bethesda consensus document (Wood et al., 2007). In the case of this T cell panel, the mAbs selected were CD45, CD2, CD3, CD4, CD5, CD7, CD8, CD56, and CD34.
The laboratory is using a 3 laser LSRII with 5 colors off the 488, 3 colors off the 640 and 2 off the 405 nm lasers. Initially, the detectors were optimized using bi-exponential displays with stained cells to define the minimal voltage for each channel as that voltage where the signal to noise ratio of stained versus unstained cells plateaued. Linearity was checked with 8 color beads. Subsequently, the voltages were calibrated daily using CS&T beads. He collects 100,000 viable events routinely and 300,000 events for MRD detection. The gating strategy starts with doublet discrimination, followed by forward scatter versus side scatter, DAPI versus side scatter to exclude dead cells, and finally CD45 versus side scatter to color code leukocyte populations.
To illustrate the process of creating and validating a new mAb cocktail, 2 case studies were presented.
Case Study 1: a sample from a 58 year old male with a t(11,14), PAX5+ malignancy was sent to flow cytometry to rule out mantle cell versus plasma cell myeloma. It was run by flow in two tubes, one for B cells and another for plasma cells. The abnormal population expressed CD45(dim), CD38, CD20, CD19(heterogeneous), surface and cytoplasmic kappa light chain and was negative for CD5, CD10 and lambda. Based on these results, it was tentatively classified as a plasma cell myeloma. In the future, however, it was desired to have a single tube to address this situation.
To create a one tube cocktail, the following combination of mAbs where selected: CD45/CD19/CD22/CD38/CD138/HLADr/CD5. The next step was to create a grid identifying validated mAbs versus the fluorochromes that were available in the laboratory. The final step in determining which fluorochromes mAb combinations were most appropriate used the following rules:
- Include CD45 as a backbone mAb in a fluorochrome common to other panels
- Strongly expressed antigens were placed on the dimer fluorochromes
- Selected IgG1 mAbs over IgG2 mAbs, because both IgG2a and IgG2b isotypes interact with compliment in unwashed blood (Wood and Levin, 2006).
Once a tentative cocktail was chosen it was validated by running on 10 healthy donor samples, 10 bone marrow samples from patients with abnormal B cell populations and 10 bone marrow samples from patients with abnormal plasma cells. Dr. Kussick stressed that it’s important to correlate the flow results from the validation study with other information from the patient’s immunohistochemistry tests and the clinical findings.
The final configuration selected was DAPI/CD45 V500/CD52 FITC/CD22 PE/CD19 PE-TxRed/CD138 PE-Cy7/CD5 APC/CD38 APC-Ax750. Using this cocktail the index case was CD38(bright), CD138(heterogeneous), CD19(heterogeneous), CD52(dim), CD22(-), and HLADr(heterogeneous). A similar later case was CD38(bright), CD138(bright), CD19(-), CD52(dim), CD22(-), and CD5(-).
Case Study 2 explored a situation where a mAb routinely used by the laboratory became unavailable. The lab had a panel for aplastic large cell lymphoma with CD30 in APC. When this mAb conjugate combination became unavailable, the decision was made to move CD30 PE. The final configuration was DAPI/CD45 V500/CD30 PE/CD3 PE-TxRed/CD25 PE-Cy5/HLADr PE-Cy7/ CD4 APC-Ax750. The problem with validating this panel is that very few normal cells express CD30. The solution used was to spike peripheral blood with a cell line expressing CD30. Several lines including K562 and Karpas 299 cells derived from an erythroleukemia and anaplastic large cell lymphoma, respectively, express CD30. The cocktail was then validated using the spiked blood at a 10:1 ratio. Included with each sample was an FMO control that replaced the CD30 with an isotype control.
In summary, when designing a panel Dr. Kussick suggests carefully considering the question(s) to be answered. If the assay is targeted at an abnormal population, identify 1, 2 or more gating antigens to localize this population, e.g. CD45/CD38 from case 1 and CD25/HLADr for case 2. Use 1 or more mAbs to separate normal populations away from the population of interest. For situations with rare positive cases for the validation, spike an appropriate cell line into normal peripheral blood or bone marrow.
4. “Design of Multicolor Antibody Panels- the EuroFlow Experience”, Jacques J. M. vanDongen, Erasmus MC, Netherlands.
The initial EuroFlow group consisted of 7 different laboratories, all regarded as experts in flow cytometry. It has grown to 15 participating facilities with complementarily expertise to cover all aspects of panel development, standardization, and validation. The EuroFlow members are working on: (A) the development of standardized 8-color antibody panels for the diagnosis and monitoring of hematological malignancies; (B) the development of new software tools to handle large data sets and for integrating 8-color flow tubes into a single multicolor data analysis; (C) the creation of a large data base with hundreds of well-defined normal, reactive and malignant cell samples to make possible automated comparison with newly analyzed patient samples.
The EuroFlow process starts off with the selection of 1 of 3 screens based on the clinical findings: the acute leukemia orientation tube (ALOT), lymphocytosis screening tube (LST), and the plasma cell dyscrasia tubes (PCD). For limited samples the small sample tube (SST) is used. These panels are listed in Table 2.
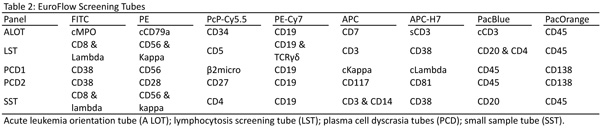
As an example of the methodical considerations, Dr. vanDongen described the process undertaken to create their tubes for chronic B-cell lymphoproliferative diseases. Initially, these patients would be screened with the LST tube. The LST was designed as a screen for several suspected clinical conditions, including lymphocytosis, lymph node enlargement, splenomegaly, unexplained cytopenias, etc. It detects aberrant mature lymphocyte populations of B, T and NK lineage. Based on the initial identification of the malignancy in this screen, the abnormal population would be further classified using the appropriate B, T, or NK-cell chronic lymphoproliferative disease panels or the plasma cell dyscrasia panel.
In this example case, a sample screened positive for a B-cell chronic lymphoproliferative disease would then be assayed in the B-CLPD panel consisting of four 8-color tubes. To initially create these tubes, 66 markers from the literature where evaluated on 150 cases of B-NHL with the goals of selecting markers which increased the differential classification of B-NHL while avoiding redundant information. These included backbone mAbs (CD45, CD19, CD20), markers for lineage assignment and maturation stage (IgM, CD10, CD38), disease specific antigens (CD5, CD23, CD79b) and integrin and chemokine receptors (CD11c, CD31, CD62L). From these, 24 markers were selected based on their ranked ability to resolve and classify the B cell malignancies. For example in CLL, IgM, CD200, CD79b, CD23 and CD38 ranked the highest, whereas in mantle cell lymphoma CD38, CD10, IgM, CD200 and CD81 were the most selective.
The backbone set of markers included in every B-CLPD tube selected was CD19, CD20 and CD45. These are not only important for combining results from different tubes but are also critical to resolve abnormal populations so the software can make comparisons to the diagnostic sample for MRD or with its database for classification of the disease. The B-CLPD panel works best when gating on the backbone markers results in at least 90% purity of the malignant B-cell population. The tubes created using the remaining markers selected based on their discriminating ability are shown in Table 3. Tubes 2 and 3 contain the most informative markers for B-NHL classification and on their own allow the classification of most typical mature B-cell malignancies (e.g. CLL, HCL). Tubes 4 and 5 are useful to distinguish immunophenotypically similar diseases from each other (e.g. DLBCL, LPL, MZL) or for further clarification of atypical cases.
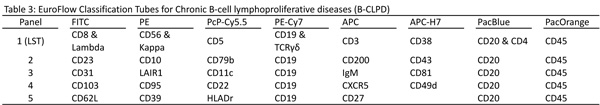
In many situations, the LST tube will clarify how difficult a newly diagnosed B-NHL will be to classify (i.e. a light chain restricted CD19+, CD20(dim), CD5+ is most likely a CLL or possibly a mantle cell). If after screening the probability of a particular B-NHL is high then a limited number of B-CLPD tubes sufficient to identify with high probability the abnormal population are run. More difficult cases require the full panel.
Dr. vanDongen also described how the Infinicyt software's automated pattern-guided principal component analysis (PCA) approach was able to appropriately separate and classify B-NHL. Details of this approach, using a 4 color data set, are described by (Costa et al., 2010). More information about the Infinicyt software can be found at http://www.infinicyt.com.
In summary, the EuroFlow antibody panels were built on evidence based strategy aiming for the most informative and reproducible combinations. Fluorochromes and specific clones were tested in several testing rounds within the EuroFlow laboratories before approval. In this process, clinical relevance, technical issues and cost effective aspects were taken into account. Considerable effort also went into standardizing instrument setting and SOP’s. The result has been that using EuroFlow cocktails in conjunction with the Infinicyt software and database has enabled participating laboratories to diagnose, characterize and subclassify the major WHO entities. Moreover, the antibody panels are flexible, not all tubes are needed and laboratory specific combinations can be included.
References:
Borowitz, M.J., F.E. Craig, J.A. Digiuseppe, A.J. Illingworth, W. Rosse, D.R. Sutherland, C.T. Wittwer, and S.J. Richards. 2010. Guidelines for the diagnosis and monitoring of paroxysmal nocturnal hemoglobinuria and related disorders by flow cytometry. Cytometry 78:211-230.
Costa, E.S., C.E. Pedreira, S. Barrena, Q. Lecrevisse, J. Flores, S. Quijano, J. Almeida, M. del Carmen Garcia-Macias, S. Bottcher, J.J. Van Dongen, and A. Orfao. 2010. Automated pattern-guided principal component analysis vs expert-based immunophenotypic classification of B-cell chronic lymphoproliferative disorders: a step forward in the standardization of clinical immunophenotyping. Leukemia : official journal of the Leukemia Society of America, Leukemia Research Fund, U.K 24:1927-1933.
Wood, B.L., M. Arroz, D. Barnett, J. DiGuiseppe, B. Greig, S.J. Kussick, T. Oldaker, M. Shenkin, E. Stone, and P.K. Wallace. 2007. 2006 Bethesda international consensus recommendations on the immunophenotypic analysis of hematolymphoid neoplasia by flow cytometry: optimal reagents and reporting for the flow cytometric diagnosis of hematopoietic neoplasia. Cytometry 72 Suppl 1:S14-22.
Wood, B.L., and G.R. Levin. 2006. Interactions between mouse IgG2 antibodies are common and mediated by plasma C1q. Cytometry 70:321-328.

Paul K. Wallace, PhD
Roswell Park Cancer Institute, Buffalo, NY

