What is ALPS?
Autoimmune lymphoproliferative syndrome (ALPS), first described by Canale and Smith in 1967, is a genetic disorder of the immune system caused by defective Fas-mediated apoptosis. [1] ALPS patients have a defective Fas mediated apoptotic pathway, leading to the pathognomonic features such as nonmalignant lymphoproliferation, autoimmunity, and an increased risk of malignancies.[2] A hallmark of ALPS is increased numbers of CD4 and CD8 double negative T-cells (DNTs) in the circulation and lymphoid tissue.[3] ALPS is a rare disease but may be more common than previously thought. [4]
Pathophysiology of ALPS
The molecular basis of ALPS was elucidated in 1995 when mutations in the FAS gene were identified in patients with apoptosis defects. [5] These patients with autoimmunity, lymphoproliferation, and increased numbers of DNTs showed similar clinical phenotypes with 2 mouse models that caused autoimmune disease, lpr and gld, which are due to recessive mutations in genes encoding Fas (CD95, Apo-1) and FasL (CD95L), respectively, leading to defective Fas mediated apoptosis. Based on these similarities, these patients were then classified as having ALPS.[3, 6]
Fas is a cell surface receptor belonging to the tumor necrosis factor receptor superfamily. [6] Fas is normally highly expressed in activated B and T cells.[7] Fas, when engaged by its ligand (Fas ligand), initiates a cascade of intracellular events leading to activation of caspase 8 and caspase 10 proteases, which ultimately lead to apoptosis of the cell. [7, 8] The Fas apoptotic pathway is critical for the down regulation of the immune response by inducing apoptosis of excess activated, antigen driven or autoreactive cells. Mutations in the Fas pathway lead to lymphoid hyperplasia and autoimmunity consequent to abnormal lymphocyte survival. The lymphoproliferation that results is from the accumulation of lymphocytes that have not undergone normal apoptosis. ALPS patients exhibit a TH2 cytokine profile that is thought to favor antibody production and thus lead to the autoimmune manifestations seen in ALPS.[9]
What are DNT cells?
DNT cells are a consistent finding in ALPS. DNT cells are found in normal individuals in small amounts but can reach up to 40-60% in ALPS patients.[5, 10] These DNT cells express alpha beta TCR chains, coexpress the CD45RA isoform B220, and are high producers of IL-10 in contrast to DNT cells from normal individuals. [10] [11]The definitive origin of DNT cells still remains undetermined. Some recent literature supports DNT cells being of CD8 origin, while others they believe they are activated mature T cells that have lost CD4 or CD8 coreceptor expression, or a minor cell lineage that is expanded in the setting of defective FAS signaling.[9]
Molecular basis of ALPS
ALPS is caused by mutations in the Fas gene and other genes in the Fas signaling pathway such as the Fas molecule, its ligand or in caspase -10, caspase-8 or the Fas-associated death domain (FADD). The mutations in the different Fas signaling genes has led to the classification of ALPS into different subgroups: ALPS-0 have homozygous mutations in the Fas gene, Ia has heterozygous mutations in the Fas gene, Ib has mutations in the gene encoding Fas ligand(FAS-L); Alps IIa and IIb with mutations in CASPASE-10 and CASPASE 8 genes, respectively; ALPS III with unknown genetic defects but with the ALPS clinical phenotype, ALPS IV with NRAS mutations, and finally ALPS Im with somatic mosaic FAS mutations.[12-14] [15]
How does ALPS present?
ALPS typically presents in childhood with lymphadenopathy, which can be massive, organomegaly, autoimmunity, most commonly in the form of autoimmune cytopenias, and lymphocytosis. While clinical manifestations usually occur in pediatric patients with an average age of 22 months at the time of diagnosis, patients may present into adolescence and early adulthood. In many ALPS patients, the symptoms may improve with age but the cytopenias often show a relapsing course and the lymphoproliferation tends to continue, accounting for the high risk of developing malignancies, especially lymphoma. The risk for developing lymphoma is as high as 51 times greater than normal for Hodgkin lymphoma and 14 times greater than normal for non-Hodgkin lymphoma.
How is ALPS diagnosed?
The NIH established initial criteria to diagnose ALPS in 1999. Since then, there have been significant advances in the understanding of the disease. Recently, the diagnostic criteria have been revised in order to simplify the diagnosis and classification of ALPS.[16] In addition, the revised criteria addressed potential problems that may have hindered the use of the previous classification system. For instance, the lymphocyte proliferation assay is a resource intensive test that is only offered at select centers and is not standardized among centers leading to variable results. New studies have shown that new biomarkers have been able to predict ALPS.[16]
Currently, in the revised diagnostic criteria for ALPS there are two required criteria and accessory criteria that are subdivided into primary and secondary accessory categories. The two required criteria that must be met for a diagnosis of ALPS include: first, chronic, nonmalignant or noninfectious lymphadenopathy and/or splenomegaly; second, elevated DNT cells that are CD3+, TCR alpha beta +, CD4-,CD8- DNT cells that are >1.5% of total lymphocytes or >2.5% of CD3+ lymphocytes. [16]
The accessory criteria include two primary accessory criteria and four secondary accessory criteria. The primary accessory criteria include: 1) defective lymphocyte apoptosis, 2) somatic or germline pathogenic mutation in FAS< FASLG, or CASP10. The secondary accessory criteria include 1) elevated plasma s FASL levels or elevated plasma IL-10 levels or elevated plasma or serum vitamin B12 levels, or elevated plasma IL-18 levels, 2) typical immunohistological findings, 3) autoimmune cytopenias AND elevated IgG levels, and 4) family history of nonmalignant/noninfectious lymphoproliferation with or without autoimmunity. [16]
A definitive diagnosis of ALPS is based on the presence of both required criteria plus one primary accessory criterion. A probable diagnosis of ALPS is based on the presence of both required criteria plus one secondary accessory criterion. (see Table 1) [16]
Table 1
Revised diagnostic criteria for ALPS
Required
1. Chronic(>6 months), nonmalignant, noninfectious lymphadenopathy or splenomegaly or both
2. Elevated CD3+ TCRalphabeta+CD4-CD8- DNT cells (> 1.5% of total lymphocytes or 2.5% ofCD3+ lymphocytes) in the setting of normal or elevated lymphocyte counts
Accessory
Primary
- Defective lymphocyte apoptosis (in 2 separate assays)
- Somatic or germline pathogenic mutation in FAS, FASL, or CASP10
Secondary
- Elevated plasma sFASL levels (>200pg/ml) OR elevated plasma interleukin-10 levels (>20 pg/ml) OR elevated serumor plasma vitamin B 12 levels (>1500 ng/L) OR elevated plasma interleukin -18 levels >500 pg/ml
- Typical immunohistological findings as reviewed by an experienced hematopathologist
- Autoimmune cytopenias (hemolytic anemia, thrombocytopenia or neutropenia) AND elevated immunoglobulin G levels (polyclonal hypergammaglobulinemia)
- Family history of a nonmalignant/noninfectious lymphoproliferation with or without autoimmunity
A definitive diagnosis is based on the presence of both required criteria plus one primary accessory criterion. A probable diagnosis is based on the presence of both required criteria plus on secondary criterion.
(Table from reference 16 – Revised Diagnostic Criteria and Classification for ALPS, 2010)
Laboratory findings in ALPS
There are several laboratory findings that are well described in ALPS patients such as hypergammaglobulinemia, elevated plasma levels of IL-10 protein, decreased plasma levels of soluble FAS-L, and elevated serum vitamin B 12. However, the hallmark of ALPS is the expansion of a unique population of T cells that are alpha beta T cell receptor positive, as well as negative for both CD4 and CD8 coreceptors, these cells are referred to as double negative T cells (DNT cells).
Other markers may be seen on these DNT cells in ALPS patients such as an unusual B cell specific CD45R isoform, B220.[17] The coexpression of B220 on DNT cells adds significant specificity marker for ALPS and has been associated with FAS mutations. [10, 17] Another population of cells that has been described in ALPS patients is CD5 positive B cells, although this is less helpful.
Flow Cytometric Analysis for ALPS
In the Clinical Immunology Lab at The Children’s Hospital of Philadelphia, DNT cells are identified using a simple flow cytometric assay with a 2 tube panel. The first tube includes IgG1FITC, IgG1 RD1, CD3 ECD; the second tube includes TCRab FITC, CD4 RD1, CD8RD1, and CD3 ECD. These monoclones are added to 100ul of the patient’s whole blood. The tubes incubate for 15 min at 2-8C, red blood cells are lysed on a Beckman Coulter TQ- Prep and analyzed on a Beckman Coulter FC500. A CD3 versus sidescatter plot is used to gate CD3+ lymphocytes (Figure 1 and Figure 2). From this gate CD3+ lymphocytes are analyzed and a percentage of TCR alpha beta+ and CD4 and CD8 double negative T cells are calculated as a percentage of CD3 positive T cells (see Figure 1). An absolute count is also provided.
Figure 1


Figure 1 legend:
This is a normal control. This is a CD3 versus sidescatter plot (top) that is used to gate CD3+ lymphocytes. From this gate, the CD3+ lymphocytes are analyzed and a percentage of TCR alpha beta+ AND CD4 and CD8- DNTcells(bottom) are calculated as a percentage of CD3+ T cells (quadrant B4). In this control, there is 0.7% TCR alpha beta+, CD4-, CD8- DNT cells. The normal range for DNT cells at the Children’s Hospital of Philadelphia is 2.7% of CD3 positive lymphocytes. An absolute value is also given.
Interpretation of DNT cells
The presence of increased numbers of DNT cells is a requirement for the diagnosis of ALPS, as clearly stated in the diagnostic criteria. However, one must be aware that the sole finding of increased DNT cells is not diagnostic of ALPS. Moreover, it is imperative that this population of DNT cells be identified correctly and accurately, as CD3+ cells that are negative for CD4 and CD8 coexpression is not sufficient. The population of DNT cells in ALPS must be clearly distinguished from TCR gamma delta+ DNT cells by costaining with alpha beta TCR directed antibodies in the flow cytometric assay (see Figure 2). This is important because normal delta gamma TCR+ cells, which are usually a small subset of T cells in the peripheral blood, are negative for CD4 and CD8 and if they are not distinguished from the TCR alpha beta+ DNT cells they may cause a false elevation of this population.
Figure 2


Figure 2 legend:
This is a peripheral blood sample from a newly diagnosed patient with ALPS. The top plot shows the CD3 versus sidescatter plot that is used to gate on the CD3+ lymphocytes. The bottom plot shows the percentage of TCR alpha beta+ AND CD4- and CD8- T cells (DNT cells). The percentage of DNT cells in this patient is 16.2% at diagnosis. The normal range for DNT cells at the Children’s Hospital of Philadelphia is 2.7% of CD3 positive lymphocytes.
The normal value for the percentage of DNT cells in our lab at Children’s Hospital of Philadelphia is < or equal to 2.7% of CD3+ lymphocytes based on evaluation of controls during test validation. This value is slightly higher than that described by in the original criteria by the NIH, in which the cutoff for TCR alpha beta + DNT cells was 1% of total lymphocytes. In the revised diagnostic criteria, the cutoff of elevated DNT cells is 1.5% of total lymphocytes or 2.5% of CD3+ lymphocytes. Therefore, one should be aware that the cutoff for TCR alpha beta+ DNT cells is laboratory dependent and varies between laboratories; therefore, the cutoff described by the NIH may not apply to all laboratories.
The presence of increased numbers of DNT cells is one of the requirements for the diagnosis of ALPS in the appropriate clinical setting. Other criteria must be met and the diagnosis of ALPS is not based solely on the presence of increased DNT cells as other diseases such as autoimmune disorders may show increased TCR alpha beta+ DNT cells but lack the clinical picture and other criteria for ALPS. Conversely, other disorders, such as some immunologic as well as autoimmune disorders, may have a clinical presentation similar to ALPS but lack DNT cells.
If an expanded population of DNT cells is detected, this value needs to be correlated with the clinical history and symptoms (ie lymphadenopathy, autoimmune and/or histologic findings) and additional testing is indicated such as the lymphocyte apoptosis assay, genetic testing or other laboratory data such as levels of soluble FASL, vitamin B12, IL-10, or immunoglobulin G levels (see Table 1).
Laboratory experience
The Clinical Immunology Lab at the Children’s Hospital of Philadelphia performs approximately 5-10 ALPS panels per month. Of these samples, approximately 50% demonstrate an expanded number of DNT cells. In these patients, further laboratory testing is performed to confirm the diagnosis of ALPS.
How is ALPS treated?
The clinical course of ALPS shows a great deal of variability. Some patients with ALPS require no treatment, but the majority requires immunosuppressive medications, including corticosteroids. Some patients with severe, refractory cytopenias often require more aggressive immunosuppression. [4] Rapamycin (Sirolimus) is an immunosuppressive agent that targets the mammalian target of rapamycin (mTOR) and induces apoptosis in normal and abnormal lymphocytes. Rapamycin treatment has been successful in treating patients with refractory ALPS. [4, 18]
How is ALPS monitored?
ALPS patients often require immunosuppression. Immunosuppressive medications often improve lymphadenopathy and cytopenias in the majority of patients. DNT cells can be used as a biomarker of disease and can be followed longitudinally in patients using the ALPS flow cytometric panel. Peripheral blood DNT cells can be compared before and after immunosuppression. In patients on immunosuppression the population of DNT cells often is reduced (on average a decrease of 60% is seen) and may even normalize. (see Figure 3)[4]
Figure 3

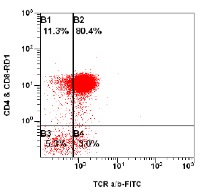
Figure 3 legend:
This is the same ALPS patient as seen in Figure 2 after Rapamycin therapy. The top plot shows the CD3 versus sidescatter plot that is used to gate CD3+ lymphocytes. The bottom plot is the CD3+ lymphocytes that are analyzed as a percentageof TCR alpha beta+, CD4-, CD8- T cells (DNT cells). This patient’s population of DNT cells decreased to 3.0% (quadrant B4) of CD3+ lymphocytes (from 16.7% at diagnosis) after Rapamycin therapy. The normal range for DNT cells at the Children’s Hospital of Philadelphia is 2.7% of CD3 positive lymphocytes.

Michele E. Paessler, D.O.
Medical Director Clinical Immunology Lab
Children’s Hospital of Philadelphia
Assistant Professor, Dept of Pathology and Laboratory Medicine
University of Pennsylvania School of Medicine
Philadelphia, PA
REFERENCES
1. Canale, V.C. and C.H. Smith, Chronic lymphadenopathy simulating malignant lymphoma. J Pediatr, 1967. 70(6): p. 891-9.
2. Bleesing, J.J., S.E. Straus, and T.A. Fleisher, Autoimmune lymphoproliferative syndrome. A human disorder of abnormal lymphocyte survival. Pediatr Clin North Am, 2000. 47(6): p. 1291-310.
3. Sneller, M.C., S.E. Straus, E.S. Jaffe, J.S. Jaffe, T.A. Fleisher, M. Stetler-Stevenson, and W. Strober, A novel lymphoproliferative/autoimmune syndrome resembling murine lpr/gld disease. J Clin Invest, 1992. 90(2): p. 334-41.
4. Teachey, D.T., D.A. Obzut, K. Axsom, J.K. Choi, K.C. Goldsmith, J. Hall, J. Hulitt, C.S. Manno, J.M. Maris, N. Rhodin, K.E. Sullivan, V.I. Brown, and S.A. Grupp, Rapamycin improves lymphoproliferative disease in murine autoimmune lymphoproliferative syndrome (ALPS). Blood, 2006. 108(6): p. 1965-71.
5. Rieux-Laucat, F., F. Le Deist, C. Hivroz, I.A. Roberts, K.M. Debatin, A. Fischer, and J.P. de Villartay, Mutations in Fas associated with human lymphoproliferative syndrome and autoimmunity. Science, 1995. 268(5215): p. 1347-9.
6. Watanabe-Fukunaga, R., C.I. Brannan, N.G. Copeland, N.A. Jenkins, and S. Nagata, Lymphoproliferation disorder in mice explained by defects in Fas antigen that mediates apoptosis. Nature, 1992. 356(6367): p. 314-7.
7. Nagata, S. and P. Golstein, The Fas death factor. Science, 1995. 267(5203): p. 1449-56.
8. Krammer, P.H., CD95's deadly mission in the immune system. Nature, 2000. 407(6805): p. 789-95.
9. Bristeau-Leprince, A., V. Mateo, A. Lim, A. Magerus-Chatinet, E. Solary, A. Fischer, F. Rieux-Laucat, and M.L. Gougeon, Human TCR alpha/beta+ CD4-CD8- double-negative T cells in patients with autoimmune lymphoproliferative syndrome express restricted Vbeta TCR diversity and are clonally related to CD8+ T cells. J Immunol, 2008. 181(1): p. 440-8.
10. Bleesing, J.J., M.R. Brown, J.K. Dale, S.E. Straus, M.J. Lenardo, J.M. Puck, T.P. Atkinson, and T.A. Fleisher, TcR-alpha/beta(+) CD4(-)CD8(-) T cells in humans with the autoimmune lymphoproliferative syndrome express a novel CD45 isoform that is analogous to murine B220 and represents a marker of altered O-glycan biosynthesis. Clin Immunol, 2001. 100(3): p. 314-24.
11. Lopatin, U., X. Yao, R.K. Williams, J.J. Bleesing, J.K. Dale, D. Wong, J. Teruya-Feldstein, S. Fritz, M.R. Morrow, I. Fuss, M.C. Sneller, M. Raffeld, T.A. Fleisher, J.M. Puck, W. Strober, E.S. Jaffe, and S.E. Straus, Increases in circulating and lymphoid tissue interleukin-10 in autoimmune lymphoproliferative syndrome are associated with disease expression. Blood, 2001. 97(10): p. 3161-70.
12. Wu, J., J. Wilson, J. He, L. Xiang, P.H. Schur, and J.D. Mountz, Fas ligand mutation in a patient with systemic lupus erythematosus and lymphoproliferative disease. J Clin Invest, 1996. 98(5): p. 1107-13.
13. Wang, J., L. Zheng, A. Lobito, F.K. Chan, J. Dale, M. Sneller, X. Yao, J.M. Puck, S.E. Straus, and M.J. Lenardo, Inherited human Caspase 10 mutations underlie defective lymphocyte and dendritic cell apoptosis in autoimmune lymphoproliferative syndrome type II. Cell, 1999. 98(1): p. 47-58.
14. Oliveira, J.B., N. Bidere, J.E. Niemela, L. Zheng, K. Sakai, C.P. Nix, R.L. Danner, J. Barb, P.J. Munson, J.M. Puck, J. Dale, S.E. Straus, T.A. Fleisher, and M.J. Lenardo, NRAS mutation causes a human autoimmune lymphoproliferative syndrome. Proc Natl Acad Sci U S A, 2007. 104(21): p. 8953-8.
15. Rieux-Laucat, F., A. Fischer, and F.L. Deist, Cell-death signaling and human disease. Curr Opin Immunol, 2003. 15(3): p. 325-31.
16. Oliveira, J.B., J.J. Bleesing, U. Dianzani, T.A. Fleisher, E.S. Jaffe, M.J. Lenardo, F. Rieux-Laucat, R.M. Siegel, H.C. Su, D.T. Teachey, and V.K. Rao, Revised diagnostic criteria and classification for the autoimmune lymphoproliferative syndrome (ALPS): report from the 2009 NIH International Workshop. Blood, 2010. 116(14): p. e35-40.
17. Bleesing, J.J., J.E. Janik, and T.A. Fleisher, Common expression of an unusual CD45 isoform on T cells from patients with large granular lymphocyte leukaemia and autoimmune lymphoproliferative syndrome. Br J Haematol, 2003. 120(1): p. 93-6.
18. Janic, M.D., C.D. Brasanac, J.S. Jankovic, B.L. Dokmanovic, R.N. Krstovski, and J.N. Kraguljac Kurtovic, Rapid regression of lymphadenopathy upon rapamycin treatment in a child with autoimmune lymphoproliferative syndrome. Pediatr Blood Cancer, 2009. 53(6): p. 1117-9.

