|
|
|
|
|
Dihydrorhodamine (DHR) flow cytometry test for Chronic Granulomatous Disease (CGD): A simple test for routine clinical flow cytometry
Chronic granulomatous disease (CGD) is an inherited disorder of phagocytes resulting in recurrent life threatening pyogenic infections and widespread granulomatous inflammation in tissue. It was first described in the 1950s as “a fatal granulomatous disease of childhood.”1 Initially, it was thought to be only an X-linked genetic disease, however, it is now known to occur in both the X-linked and autosomal recessive forms and is caused by a large heterogeneous spectrum of genetic mutations. It occurs in approximately 1:200,000 births worldwide; however, this may be considerably higher in those populations where consanguineous marriage practices occur (reviewed in 2). The pathogenesis of CGD involves a defect in the phagocytic nicotinamide dinucleotide phosphate (NADPH) oxidase complex. This defect results in lack, or reduced amount, of superoxide formation and subsequently lack, or reduced amount, of microbicidal reactive oxygen species (ROS)(hydrogen peroxide, hydroxyl radicals, peroxynitrite anions, and oxyhalides) being produced during the normal oxidative burst in response to inflammatory stimuli and phagocytosis. Consequently, the phagocytes have an inability to kill certain pathogens yielding the characteristic recurrent and severe infections seen in CGD patients. The indirect detection of these reduced levels of ROS, specifically hydrogen peroxide, is the basis of the dihydrorhodamine (DHR) flow cytometry test for CGD.
The NADPH oxidase complex is composed of 5 subunit proteins (2 membrane and 3 cytosolic components). The two membrane components, glycoprotein 91 (gp91 phox) and protein 22 (p22 phox) are bound together in a heterodimer called flavocytochrome b558. The three cytosolic protein subunits are p47 phox, p67 phox, and p40 phox. Upon appropriate activation, p47 phox and p67 phox are phosphorylated and bind together, and then with p40 phox and rac (a regulatory GTPase), all components bind to the flavocytochrome b558 to complete the active NADPH oxidase complex.3 Many genetic mutations in the genes coding for gp91 phox, p22 phox, P47 phox and p67 phox have been described which result in CGD. The most common occur in the CYBB gene located on the X chromosome which encodes for gp91 phox (also referred to as NOX2). Approximately 60% or more of all CGD patients are X-linked and demonstrate various mutations in the CYBB gene resulting in a complete lack, reduced, or non-functional gp91 phox . The next most common mutation is that which occurs in the NCF1 gene located on chromosome 7 which encodes for p47 phox and is inherited in an autosomal recessive manor. This accounts for approximately 30% of CGD patients. Mutations in the CYBA gene (on chromosome 16) encoding p22 phox and the NCF2 gene (on chromosome 1) encoding p67 phox are each responsible for approximately 5% of CGD patients and are also autosomal recessive.(reviewed in 4) Recently, a patient with chronic granulomatous inflammation of the gastrointestinal tract was described with mutations in the NCF4 gene (on chromosome 22) which encodes the p40 phox.5 Thus, defects in all five subunits of the NADPH oxidase complex have now been described resulting in deficient production of ROS in response to stimulatory signals, which the DHR flow cytometry assay can detect via reduced oxidation of the dihydrorhodamine.
The most common laboratory screening test for CGD has been the Nitroblue tetrazolium dye reduction test (NBT). There are several versions of this test, in which the dye in the presence of the oxidative burst products is reduced to visibly blue formazan. However, this test can be insensitive, is subjective, and laborious. While it can be argued that the NBT test still has a place in resource poor environments, it is generally agreed that the DHR flow cytometry test is the method of choice.6,7 The rarity of CGD has most likely influenced or precluded many clinical cytometry laboratories from thinking about or attempting to perform and bring the DHR test on line. However, this test is extremely robust, easy to perform, and inexpensive. What is more, most of the test specific reagents can be aliquoted into small single run amounts and frozen (eliminating outdating). This makes it ideal for low volume and/or infrequent testing. It is a whole blood assay that can be performed on a total of 300µl of blood per patient and if necessary can even be scaled down to 60 µl total, or less. The procedure used in our Clinical Flow Laboratory is basically that described by O’Gorman and Corrochano8 with modifications. The unique reagents in this test are the dihydrorhodamine123 (DHR123) (Molecular Probes, Eugene, OR) and phorbol 12-myristate 13-acetate (PMA) (Sigma-Aldrich, St Louis, MO). In making stock solutions, these reagents are diluted to 2500 µg/ml and 100 µg/ml, respectively, in dimethyl sulfoxide (DMSO). These stocks are then aliquoted in 35 µl and 30 µl amounts in multiple mini vials and stored at -20 degrees C until use (one mini vial of each is required per assay run). For the assay, one of each mini vial is diluted with phosphate buffered saline with azide (PBA)(Ca and Mg free with 0.2% bovine serum albumin and 0.2% sodium azide) to 15 µg/ml and 300ng/ml, respectively.
Three tubes are set up for each patient and control:
1. Blood only
2. Blood + DHR123
3. Blood + DHR123 + PMA
To all tubes 100 µl of heparinized blood is diluted 1:10 with PBA. 25 µl of thawed and diluted DHR123 (375 ng/ml final concentration) is added to tubes 2 and 3. All tubes are incubated in a 37 degrees C water bath for 15 minutes. This allows for the DHR123 to be loaded into the cells. Following this incubation, 100ul of the prepared PMA solution (30ng/ml final concentration) is added to tube 3. and all tubes are incubated an additional 15 minutes at 37 degrees C. This step allows the neutrophils to be stimulated to undergo the oxidative burst thereby oxidizing the DHR123 to its resonance form (rhodamine) which is highly green fluorescent when exposed to the 488 nm laser. After washing and centrifugation, the samples are lysed with ammonium chloride (Pharm Lyse, Becton Dickinson, Mountainview, CA) for 10 minutes in the dark, followed by centrifugation, washing, and fixing in 1% formalin. The tubes are then analyzed using the 488nm laser and the FITC filter set up. Fluorescence is quantitated by mean peak channel fluorescence (MPC-FL). Results are expressed as neutrophil oxidative index (NOI) which is the ratio of MPC-FL (PMA stimulated) over MPC-FL (unstimulated). While the granulocytes are the cells of interest (gating FSC vs SSC), gating on the monocytes is good for a low-level control and gating on the lymphocytes serves as a negative control. In addition, to these gating control strategies, a normal healthy control is also run for each assay. The DHR Flow Cytometry test can detect CGD patients, carriers, and can suggest the genotype of the CGD patients. Two cases are presented below to demonstrate typical results found in this disease using the DHR assay.
Case 1
Two four week old male identical twins presented with difficult breathing and severe pneumonia for second time since birth. The physical exam revealed that the patients were febrile and irritable, and exhibited lymphadenopathy and hepatosplenomegaly. T, B, and NK cell quantitation were normal. Immunoglobulin levels were also normal. Aspergillus was isolated from the bronchial lavage. The family history revealed an older male sibling (3 years old at the time) who also had recurrent infections including pneumonia as an infant, but seemed to be controlled presently. The two twins’ and healthy control’s DHR assay results are shown in figure 1. Neutrophils were gated (FSC vs SSC) and the results expressed in single parameter histograms (number of gated events vs. fluorescence intensity). Row (A) demonstrates the cells loaded with dye but not stimulated with PMA for the healthy control and two twins. Row (B) demonstrates the cells loaded with dye and stimulated with PMA for the control and twins. The robust signal to noise ratio is shown in the control (NOI=1,054). The two twins demonstrated absolutely no oxidative activity (NOI=1 and 1.2).

The results for the twins’ older male sibling as well as the mother are shown in figure 2. It can be seen that the older male sibling also had no oxidative activity and CGD was never diagnosed until now. The mosaic pattern seen in the carrier state of the mother is clearly demonstrated by the bimodal distribution of activity (one defective and one normal). Neutrophils using the mutant X chromosome will be defective and those using the normal X will be normal. Depending on the degree of lyonization, the bimodal distribution may not be equal. In extreme cases where the mutant X is predominate, increased susceptibility to Aspergillus infection has been reported.9 In our case, the deficient neutrophils in the mother appear to have more activity than the patients’, but this most likely represents donation or transfer of some the of oxidative metabolites from the normal neutrophils to the deficient ones. While most X-CGD (gp91phox defect) patients (87%) have no activity in the DHR test (as in the case in of Figures 1 & 2), 17% have modest DHR activity which is more consistent with autosomal recessive p47 phox CGD.7 However, this can usually be distinguished by testing and examining the mother for the mosaic pattern of the carrier state. 7

Case 2
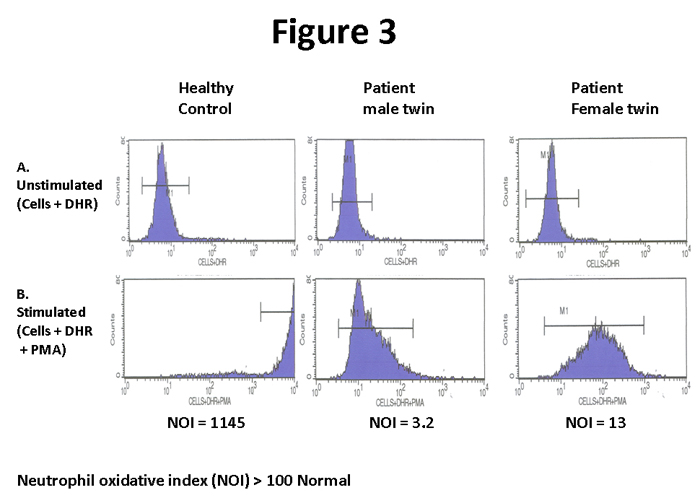
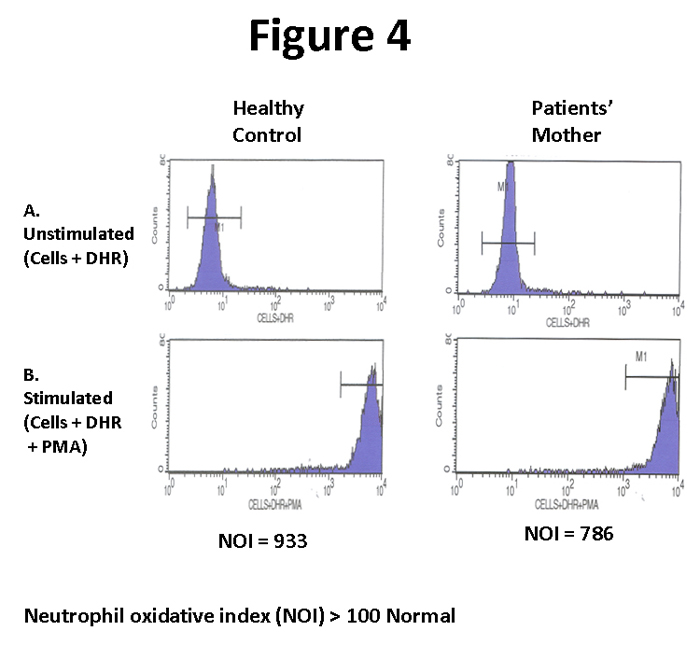
The second case is also twins, although non-identical (one male, one female). The male presented at 21 months of age with a history of recurrent infections and a severe lung abscess. Seven months later, the twin sister presented with a neck abscess. The patients were from the Middle East and a consanguineous family history was documented. In countries where consanguineous marriages are high, the autosomal recessive p47 phox CGD is much more frequent than X-linked CGD.10 Like the first case, the traditional immunological work up was non-contributory and a workup for CGD was initiated. Figure 3 demonstrates the DHR assay for the twins and the control. The assay demonstrated some activity, although markedly reduced, for both twins (NOI= 3.2 and 13, respectively). In addition, the peaks of activity are very broad in contrast to the sharp peaks found in X-linked CGD (figure 1). This is characteristic of autosomal recessive p47 phox CGD.7 However, caution must be utilized using this criterion, since the minor subset of X-linked CGD with modest activity may also have broad histograms7. Therefore, as stated above, correlation with the mother’s results is required to exclude this possibility. In this case, the mother demonstrated normal oxidative activity and not a bimodal pattern of activity (Figure 4). These patients were later confirmed by further testing to have autosomal recessive p47 phox CGD.
While the DHR assay is an extremely easy and robust assay, there are a few cautionary notes. Complete myeloperoxidase (MPO) deficiency also will cause reduced activity in the DHR test. However, MPO deficiency is generally considered to be innocuous11 and thus clinical presentation is an important consideration. In the many years that we have been testing for CGD, this has never been a problem, most likely, for the above reason. If MPO deficiency is a consideration, it can be excluded by examination of eosinophils.11 Eosinophils in MPO deficiency will exhibit normal DHR activity while in CGD, the eosinophils will be deficient.11 The more important cautionary note, in my opinion, is in patients with severe septicemia. Although rare, to date we have seen two patients with severe septicemia that exhibited a bimodal carrier state histogram. The first patient was male, so a carrier state explanation is not possible. This finding was likely due to in vivo degranulation of the neutrophils and was evident by the lower side scatter of the patient’s neutrophils compared to the control’s neutrophils. In addition, carriers of CGD are generally asymptomatic, so the flow cytometric results would not match the clinical findings even if the patient was female. Finally, how the specimens are handled can potentially cause some issues. We have noticed that older or shipped specimens may, on occasion, exhibit an elevation in the control tube (with DHR but without activation with PMA). While this results in the NOI being reduced, it is because the unstimulated tube is pre-activated and not because the stimulated tube is deficient or decreased. This is not a characteristic pattern seen in CGD and, in our hands, the ROI has not been decreased enough to make the test positive for CGD. However, to be on the conservative side, we request another specimen when this is observed.
In summary, the flow cytometry DHR test is simple, inexpensive, easy to perform, and robust. Basically, it can be perform by any routine clinical flow cytometry laboratory.

Marc Golightly, Ph.D.
Professor of Pathology
Head of Clinical Immunology and Flow Cytometry
Stony Brook University School of Medicine
Stony Brook, NY
References
1. Berendes H., Bridges, RA., and RA Good. A fatal granulomatosus of childhood: the clinical study of a new syndrome. Minn Med. 1957 May;40(5):309-12.
2. Wolach B, et al. Chronic Granulomatous disease in Israel: Clinical, functional and molecular studies of 38 patients. 2008. Clin Immunol 129:103-114.
3. Holland, SM. Chronic Granulomatous Disease. 2010. Clinic Rev Allerg Immunol 38:3-10.
4. Stasia MJ. and XJ Li. Genetics and immunopathology of chronic granulomatous disease. 2008. Semin Immunopathol 30:209-235.
5. Matute, JD., et. al. A new genetic subgroup of chronic granulomatous disease with autosomal recessive mutations in p40phox and selective defects in neutrophil NADPH oxidase activity. 2009. Blood 114(15):3309-3315.
6. Vowells, SA, Fleisher, TA., and Malech, HL. Testing for chronic granulomatous disease. 1996. Lancet 347:1048-1049.
7. Jirapongsananuruk, O., Malech, HL., Kuhns, DB., et.al. Diagnostic paradigm for evaluation of male patients with chronic granulomatous disease, based on the dihydrorhodamine 123 assay. 2003. J Allergy Clin Immunol 111(2):374-379.
8. O’Gorman, MRG. and V Corrochano. Rapid Whole-Blood Flow Cytometry Assay for the Diagnosis of Chronic Granulomatous Disease. 1995. Clin Diagn Lab Immunol 2(2): 227-232
9. Rosen-Wolff, A., et.al. Increased susceptibility of a carrier of X-linked chronic granulomatous disease (CGD) to Aspergillus fumigates infection associated with age-related skewing of lionization. 2001. Ann Hematol 80:113-115.
10. Teimourian, S., deBoer, M., and D. Roos. Molecular basis of autosomal recessive chronic granulomatous disease in Iran. 2010. J Clin Immunol 30:587-592.
11. Mauch, L., et. al. Chronic granulomatous disease (CGD) and complete Myeloperoxidase deficiency both yield strongly reduced dihydrorhodamine 123 test signals but can easily be discerned in routine testing forCGD. 2007. Clin Chem 53(5):890-896.
|
|
|

